Some viruses affect us humans, like the now all-too-familiar coronavirus SARS-CoV-2, while others mainly affect animals, such as pestiviruses that can infect pigs, cows, and even wild animals. And when farm animals get sick, it also affects farmers, food supplies, and the economy. While effective diagnostics and vaccines are available for SARS-CoV-2 due to extensive research, this is not the case for many other pathogenic viruses. To fight viruses, we need to understand how they “live” and multiply. Viruses cannot do this on their own, they use host cells to copy themselves (replication) and to build proteins (translation). And all of this is controlled by the virus’s genetic blueprint, which not only encodes proteins but also folds into special shapes, called RNA or DNA secondary structures. Just like tiny origami folding patterns hidden inside the virus’s genetic text. Viral genomes have been well studied for single species or in specific genome regions, but researchers have rarely studied the genomes of an entire virus group.
In a new study, we were able to puzzle together the full genome of all known Pestivirus species and analyse their conserved RNA secondary structures (Triebel et al., 2025). Members of the genus Pestivirus have a positive-sense, single-stranded RNA genome, which encodes a single polyprotein flanked by untranslated regions (UTRs; see panel A of the figure). Handling the diversity and the size of the genomic data required a specialized bioinformatics pipeline with the following steps:
- Selection of representative species/genomes from the entire Pestivirus genus
- Alignment of these genomes to create a full-genome representative of the genus
- Computational sequence analysis and prediction of conserved RNA secondary structures
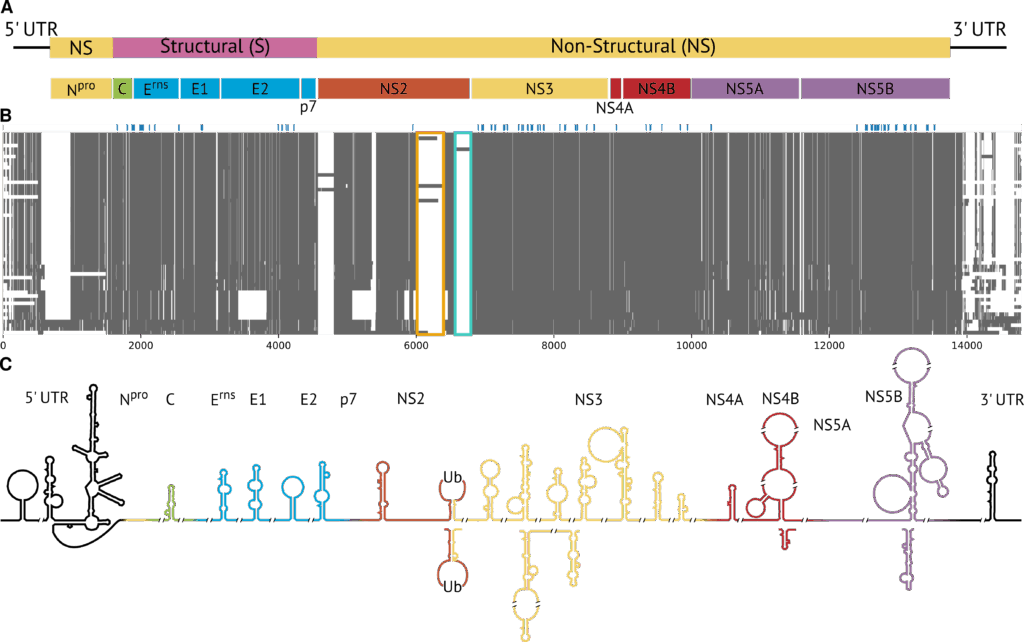
Here are the details: First, all available complete Pestivirus genomes were gathered and grouped into clusters based on how similar their sequences were. This was measured by the number of specific k-mers in the sequences using the ViralClust pipeline. For each cluster, we picked the sequence with the minimum average distance from all other genomes in the same cluster as the representative genome. This yielded 55 genomes representing all 19 known Pestivirus species.
Second, for the multiple sequence alignment, we divided the genomes into smaller, manageable pieces or “sub-alignments” using the AnchoRNA tool (described in Eulenfeld et al., 2025): Highly conserved regions present in all the sequences, called “anchors” (shown in blue in panel B of the figure), were identified and used as fixed borders between the subregions. After aligning each subregion individually, the subalignments were merged back into a single, genome-wide alignment of approximately 15,000 nucleotides for further analysis.
In the third step, we had a closer look at the sequence and secondary structures of the full Pestivirus genome. It consists of 5’ and 3’ UTRs, which are not translated into proteins, and a single open reading frame (ORF) that encodes a large polyprotein. Proteases process this polyprotein into the individual proteins required for the viral life cycle. The 5’ and 3’ UTRs and certain regions within the ORF form RNA secondary structures that play a role in genome stability, replication, and translation. We found 32 RNA secondary structures that were shared across all pestiviruses (panel C of the figure). Surprisingly, 29 were never described before and are located within the ORF. On the other hand, three of the structures are well-known and located in the 5’ and 3’ UTR. “It was exciting to find the well-known IRES [internal ribosome entry site] in the 5’ UTR of all studied Pestivirus genomes,” says Sandra Triebel, lead author of the study. “This showed that this feature is important for the entire genus and that our approach worked.” The researchers also discovered binding sites for the microRNAs MiR-17 and let-7 in more species than previously known. The binding of these short, non-coding RNAs is thought to influence replication, translation, and genome stability. Additionally, the predictions of possible structures of the RNA suggested that the 5’ and 3’ UTRs may interact over long distances in many pestiviruses, potentially leading to a circular genome.
This is the first time scientists have pieced together the complete genomic picture of all known pestiviruses and mapped the shapes their RNA could fold into. This information will serve as a foundation for further targeted research on experimental validation, better diagnostic tests, and maybe even antiviral drugs in the future: The “anchor regions” could be used to design PCR primers that can detect a broad spectrum of Pestivirus species with only a few primer sets. And the IRES element, microRNA binding sites, and long-range 5′–3′ UTR interaction sites could serve as promising starting points for antiviral strategies.
This bioinformatics pipeline is now also applied to other virus groups, including coronaviruses, influenza viruses, flaviviruses, and filoviruses, with the goal of uncovering common genetic features that could be turned into useful tools for therapies.
Contact: Sandra Triebel (sandra.triebel@uni-jena.de), Manja Marz (manja@uni-jena.de)
Original Publications:
Triebel, Sandra; Eulenfeld, Tom; Ontiveros-Palacios, Nancy; Sweeney, Blake; Tautz, Norbert; Marz, Manja (2025) First full-genome alignment representative for the genus Pestivirus. bioRxiv 2025.05.22.655560. doi: 10.1101/2025.05.22.655560.
Eulenfeld, Tom; Triebel, Sandra; Marz, Manja (2025) AnchoRNA: Full virus genome alignments through conserved anchor regions. bioRxiv 2025.01.30.635689. doi: 10.1101/2025.01.30.635689.
Funding: NFDI4Microbiota (NFDI 28/1), Cluster of Excellence Balance of the Microverse (EXC 2051), Landesprogramm ProDigital (DigLeben-5575/10-9)
Author: Maria Fabisch